
ISO 13485 — это международный стандарт, который описывает, как должна быть устроена система менеджмента качества в компании, работающей с медицинскими изделиями. Его задача не в том, чтобы у организации появилась «красивая папка с документами», и не только в том, чтобы получить сертификат. Главный смысл стандарта — помочь компании стабильно выпускать безопасные, соответствующие требованиям и прослеживаемые медицинские изделия на всех этапах их жизненного цикла. Подробнее об этой теме смотри в материале «Жизненный цикл медицинского изделия и требования ISO 13485».
Если говорить совсем просто, ISO 13485 — это правила управления компанией, при которых качество встроено в повседневную работу: в проектирование, закупки, производство, контроль, хранение, стерилизацию, поставку, обработку жалоб, обслуживание и внесение изменений. Стандарт нужен для того, чтобы качество не зависело только от опыта и внимательности отдельных сотрудников, а было управляемым, повторяемым и подтверждаемым записями.
Эта статья будет полезна руководителям компаний, специалистам по качеству, сотрудникам по регуляторным вопросам, производителям, контрактным производителям, поставщикам компонентов, внутренним аудиторам и тем, кто только планирует внедрение ISO 13485. Если вы находитесь в начале пути, отдельно рекомендуем материал «Как внедрить ISO 13485: пошаговый план» — он поможет понять последовательность действий от первичной диагностики до подготовки к сертификации.
Что это такое простыми словами
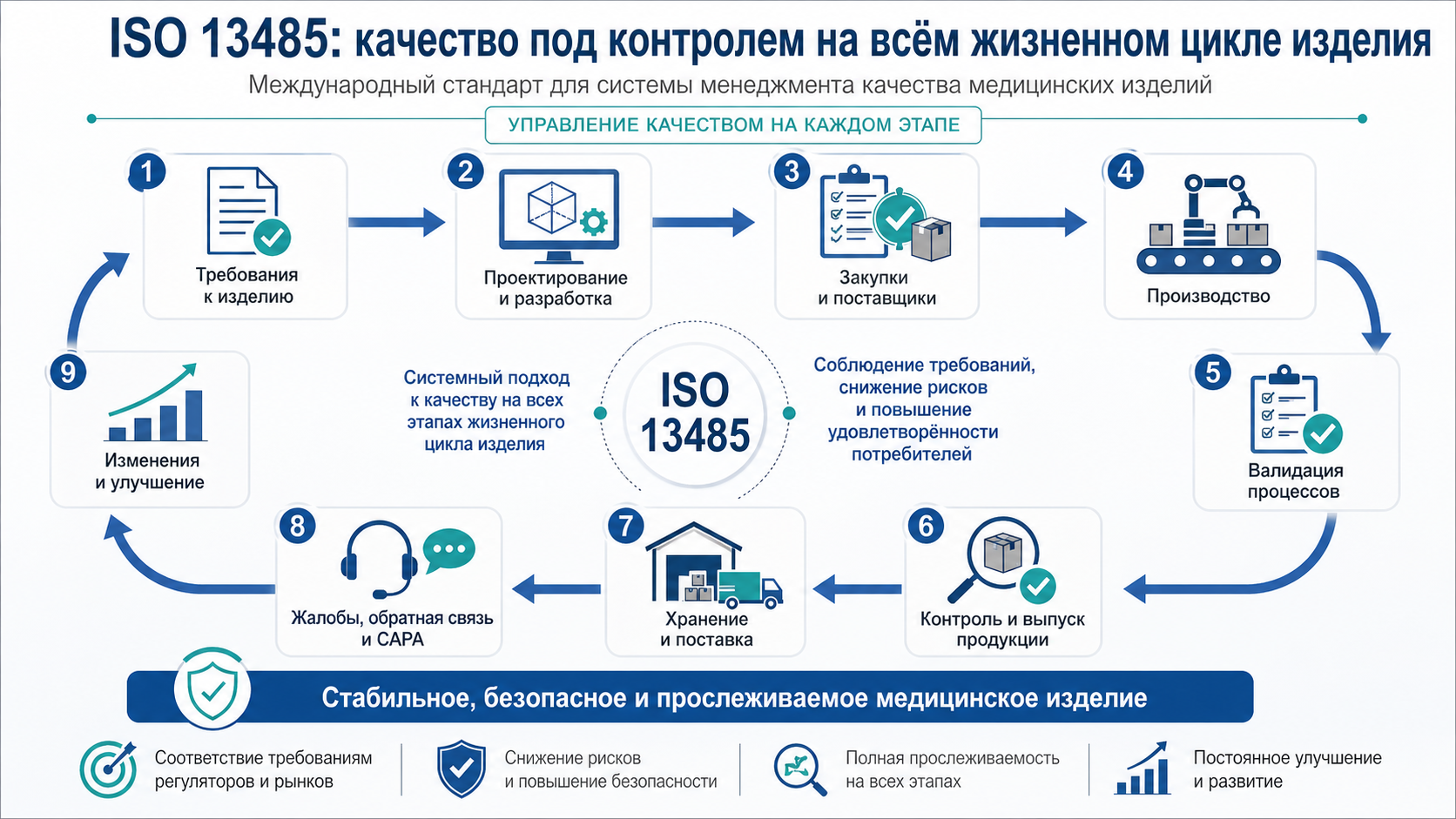
ISO 13485 — это стандарт для организаций, которые разрабатывают, производят, поставляют, обслуживают или иным образом участвуют в жизненном цикле медицинских изделий. Он устанавливает требования к тому, как компания должна управлять качеством, рисками, документированной информацией, поставщиками, несоответствиями, изменениями и обратной связью от рынка.
Обычный подход “надо делать качественно” здесь не работает. ISO 13485 переводит качество в конкретные управленческие механизмы. Компания должна заранее определить:
- кто за что отвечает;
- какие процессы влияют на безопасность и соответствие изделия;
- какие документы и записи нужно вести;
- как обеспечивается прослеживаемость;
- как контролируются изменения;
- как управлять несоответствующей продукцией;
- как устранять причины проблем, а не только их последствия;
- как подтверждать результативность процессов, которые нельзя полноценно проверить только на выходе.
Эти требования могут казаться общими, но на практике за каждым из них стоят конкретные пункты стандарта: управление процессами, ответственность руководства, ресурсы, проектирование, закупки, производство, контроль, измерения, анализ и улучшение. Если нужно разобраться не только в общей логике, но и в структуре требований, смотри отдельный материал «Требования ISO 13485 — разбор по разделам и пунктам простыми словами».
Зачем это нужно компании и бизнесу
На старте многие компании воспринимают внедрение ISO 13485 как формальное требование: без него сложнее выйти на рынок, пройти проверку партнера или подготовиться к сертификации. Но в реальности его польза намного шире.
Во-первых, стандарт делает работу более управляемой. Когда процессы проектирования, закупок, производства, выпуска продукции, контроля изменений и обработки жалоб выстроены системно, компания меньше зависит от отдельных людей и лучше понимает, что происходит внутри.
Во-вторых, ISO 13485 помогает снизить стоимость ошибок. В сфере медицинских изделий ошибка — это не только брак, списание партии или возврат. Это еще и риск для пациента, рекламации, отзыв продукции, претензии со стороны регулятора, задержка поставок, дополнительные проверки, переработка документации и репутационные потери. Один слабо управляемый процесс может обойтись бизнесу очень дорого.
В-третьих, сертификация по ISO 13485 часто повышает доверие со стороны заказчиков, дистрибьюторов, контрактных производителей и партнеров. Для многих рынков наличие работающей системы менеджмента качества — уже не преимущество, а базовое ожидание. Подробнее о том, каким компаниям особенно полезен этот стандарт, смотри в материале «Кому подходит ISO 13485 и зачем он нужен».
Наконец, стандарт помогает руководству смотреть на качество не как на задачу одного отдела, а как на часть общего управления компанией: с метриками, ответственностью, анализом данных, корректирующими действиями и регулярной оценкой того, насколько система действительно работает. Поэтому в ISO 13485 важны не только процедуры, но и управленческие ориентиры: политика в области качества, цели, ответственность и анализ результативности системы. Подробнее об этом смотри в материале «Политика и цели в области качества по ISO 13485».
Как это связано с системой менеджмента качества для медицинских изделий
ISO 13485 иногда упрощенно называют «ISO 9001 для медицинских производителей». Такое сравнение может быть полезно только на самом общем уровне, но на практике оно часто мешает. ISO 13485 устроен значительно строже и теснее связан с регуляторной логикой отрасли. В чем именно различаются эти стандарты, подробно разобрано в материале «ISO 13485 и ISO 9001: в чем разница».
Если общая система менеджмента качества может делать больший акцент на удовлетворенности клиента и общем совершенствовании процессов, то ISO 13485 гораздо сильнее сосредоточен на безопасности изделия, выполнении обязательных требований, прослеживаемости, управлении рисками, валидации процессов и доказуемости того, что компания действительно контролирует свою деятельность.
На практике система менеджмента качества медицинских изделий обычно включает:
- проектирование и разработку, если компания создает собственную продукцию;
- управление документированной информацией;
- оценку и контроль поставщиков;
- закупки и входной контроль;
- производство и контроль условий производства;
- валидацию процессов;
- идентификацию и прослеживаемость;
- выпуск продукции;
- управление несоответствующей продукцией;
- работу с жалобами, обратной связью и процессами после вывода продукции на рынок;
- корректирующие и предупреждающие действия;
- внутренние аудиты;
- анализ со стороны руководства;
- обучение и подтверждение компетентности персонала.
Зрелая система связывает все эти элементы между собой. Незрелая система хранит их как набор отдельных процедур, которые вспоминают только перед аудитом.
Какие риски, процессы и регуляторные требования важно учитывать
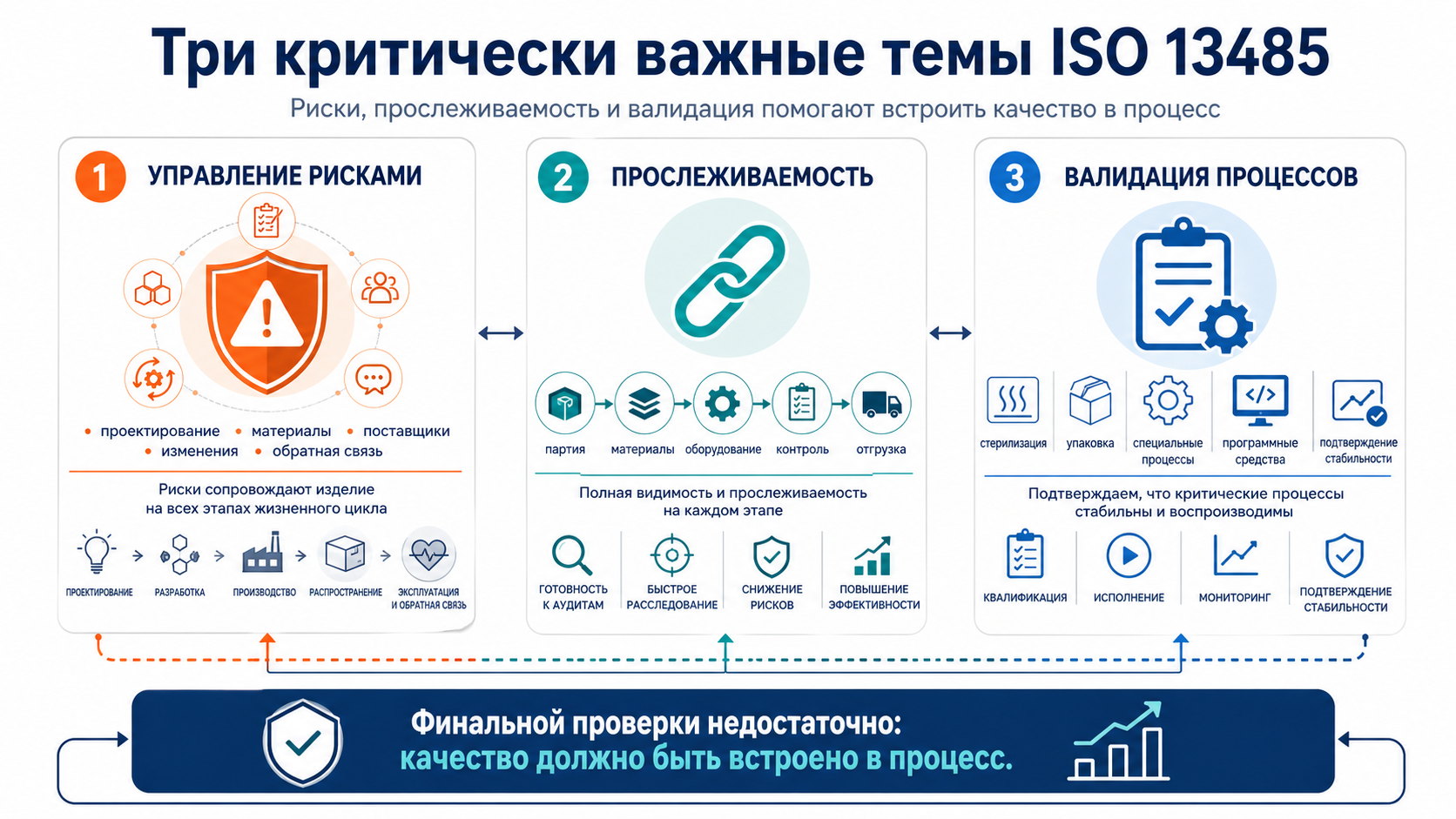
Одна из ключевых особенностей медицинских изделий в том, что качество здесь нельзя обеспечить только финальной проверкой. Если компания надеется “поймать все проблемы на выходе”, значит, она уже опоздала. Управлять нужно не только результатом, но и процессами, которые к нему приводят.
Управление рисками
Управление рисками — это не отдельный файл, который существует ради аудита. Это способ принимать решения в компании. Риски должны учитываться при проектировании, выборе материалов, закупках, смене поставщика, изменении процесса, производстве, упаковке, стерилизации, хранении и работе с обратной связью от рынка.
Например, если организация меняет поставщика важного компонента, вопрос не ограничивается ценой и сроками поставки. Нужно оценить, как это повлияет на безопасность, характеристики изделия, стабильность процесса, необходимость дополнительных испытаний и объем контрольных мероприятий. Подробнее о требованиях к риск-ориентированному подходу смотри в материале «Управление рисками в ISO 13485: требования стандарта».
Отдельно важно понимать связь ISO 13485 с ISO 14971. ISO 13485 задает требования к системе менеджмента качества, в которой риски должны учитываться при управлении процессами, продукцией и изменениями. ISO 14971, в свою очередь, подробнее раскрывает управление рисками именно для медицинских изделий. Подробнее об этой связке смотри в материале «ISO 13485 и ISO 14971: как связаны качество и управление рисками».
Прослеживаемость
Прослеживаемость — это способность восстановить историю изделия или партии: какие материалы использовались, какой поставщик их поставил, на каком оборудовании выполнялись операции, какие проверки были проведены, кто выпустил продукцию и куда она была отгружена.
Для одних видов медицинских изделий требования к прослеживаемости глубже, для других — проще. Но в любом случае именно прослеживаемость помогает компании быстро локализовать проблему, если возникла жалоба, обнаружен дефект или требуется отзыв продукции. Подробнее об этой теме смотри в материале «Прослеживаемость медицинских изделий по ISO 13485: что требует стандарт».
Валидация процессов
Есть процессы, результат которых нельзя полностью подтвердить только итоговой проверкой. Классические примеры — стерилизация, герметичная упаковка, некоторые специальные технологические операции, отдельные автоматизированные этапы и программные средства, влияющие на качество. В таких случаях особенно важно заранее доказать, что процесс стабильно дает требуемый результат. Подробнее об этом смотри в материале «Валидация процессов по ISO 13485: основные требования».
В таких случаях требуется валидация процессов. Валидация — это подтверждение того, что процесс при заданных условиях стабильно дает нужный результат. Иначе говоря, компания должна доказать не только то, что конкретная партия выглядит приемлемо, но и то, что сам процесс надежен и воспроизводим.
Связь с обязательными требованиями
ISO 13485 не заменяет обязательные регуляторные требования к медицинским изделиям. Но он помогает построить систему, через которую этими требованиями можно управлять. Если у компании слабый контроль документов, изменений, записей, поставщиков, жалоб и корректирующих действий, она будет испытывать проблемы не только на сертификационном аудите, но и в повседневной работе с обязательными требованиями рынка.
Что важно учитывать на практике
На бумаге требования ISO 13485 часто выглядят понятными. Сложности начинаются тогда, когда стандарт надо встроить в реальные процессы компании.
Проектирование и разработка
Если компания разрабатывает медицинские изделия, процесс должен быть управляемым от начала до конца: от исходных требований к изделию до проверок, анализа, подтверждения соответствия и контроля изменений. Частая ошибка — считать, что разработка заканчивается в момент вывода продукта на рынок. На практике изменения конструкции, материалов, программного обеспечения, маркировки и упаковки продолжаются, а значит, процесс должен оставаться под контролем. Подробнее об этой теме смотри в материале «Проектирование и разработка по ISO 13485: требования стандарта».
Поставщики и аутсорсинг
Управление поставщиками — это не просто перечень одобренных организаций. Компания должна понимать, какие поставщики критичны, по каким критериям они оцениваются, какие требования к ним предъявляются и как проверяется их результативность.
Особенно это важно, если часть работ передана на сторону. Когда внешний подрядчик выполняет стерилизацию, упаковку, испытания или производит важные компоненты, ответственность самой компании никуда не исчезает. Именно поэтому аудиторы обычно смотрят, как организация управляет внешними исполнителями: через договорные требования, квалификацию, входной контроль, мониторинг качества и регулярную переоценку. Подробнее об этой теме смотри в материале «Управление поставщиками и закупками по ISO 13485».
Документы и записи
Документированная информация нужна не ради бюрократии. Она нужна для двух вещей: чтобы сотрудники понимали, как именно надо работать, и чтобы компания могла доказать, что это действительно было сделано. В ISO 13485 документы и записи играют особенно важную роль, потому что именно через них подтверждается управляемость процессов, выпуск продукции, контроль изменений, работа с поставщиками, обучение, жалобы и корректирующие действия. Подробнее об этой теме смотри в материале «Документированная информация в ISO 13485: какие требования нужно учитывать».
К документированной информации относятся процедуры, инструкции, спецификации, формы записей, протоколы контроля, данные о выпуске, обучении, отклонениях, жалобах и корректирующих действиях. Но здесь важен баланс.
Слабый подход — когда документы минимальны, а ключевые знания держатся “в головах”. Другой слабый подход — когда документов слишком много, они перегружены, оторваны от реальной практики и не помогают сотрудникам в работе.
При этом важно не просто «собрать папку документов», а понять, какие процедуры, формы, записи и подтверждения действительно нужны конкретной организации. Для производителя, контрактного изготовителя, поставщика компонентов или сервисной организации набор документов может отличаться. Практический перечень можно посмотреть в материале «Какие документы нужны для ISO 13485».
Корректирующие и предупреждающие действия
В профессиональной среде часто используют сокращение CAPA (система корректирующих и предупреждающих действий). Под этим понимают работу компании по выявлению причин проблем, устранению этих причин и предупреждению повторения подобных ситуаций.
Смысл здесь не в том, чтобы формально “закрыть замечание”. Если организация сталкивается с рекламациями, внутренним браком, ошибками в документах или отклонениями в производстве и каждый раз ограничивается разовым исправлением, это незрелый подход. Зрелая система ищет корневую причину, оценивает влияние проблемы на уже выпущенную продукцию, принимает меры и затем проверяет, дали ли они реальный результат.
Типовые ошибки и слабые места

При внедрении ISO 13485 компании часто совершают похожие ошибки.
Первая ошибка — воспринимать систему качества как проект только отдела качества. Если в нее не вовлечены производство, закупки, разработка, склад, сервис и руководство, система останется формальной.
Вторая ошибка — путать наличие документов с реальной работоспособностью системы. Набор шаблонов еще не означает выполнение требований стандарта.
Третья ошибка — недооценивать контроль изменений. Поменяли поставщика, материал, этикетку, упаковку, программный модуль, метод испытаний или технологический маршрут, но не оценили последствия. Именно в таких местах часто возникают скрытые риски.
Четвертая ошибка — формально вести управление рисками. Документ есть, таблица рисков есть, но на реальные решения они не влияют. В результате управление рисками существует отдельно от закупок, производства, разработки и работы с несоответствиями.
Пятая ошибка — слабая прослеживаемость. Пока проблем нет, это может быть незаметно. Но как только приходит жалоба или требуется разобраться с конкретной партией, компания не может быстро собрать цепочку данных.
Шестая ошибка — неподготовленность к аудиту из-за слабых записей. Сотрудники могут знать “правильные слова”, но реальные записи оказываются неполными, противоречивыми или отсутствуют вовсе.
Что проверяют на аудите и на что обратить внимание
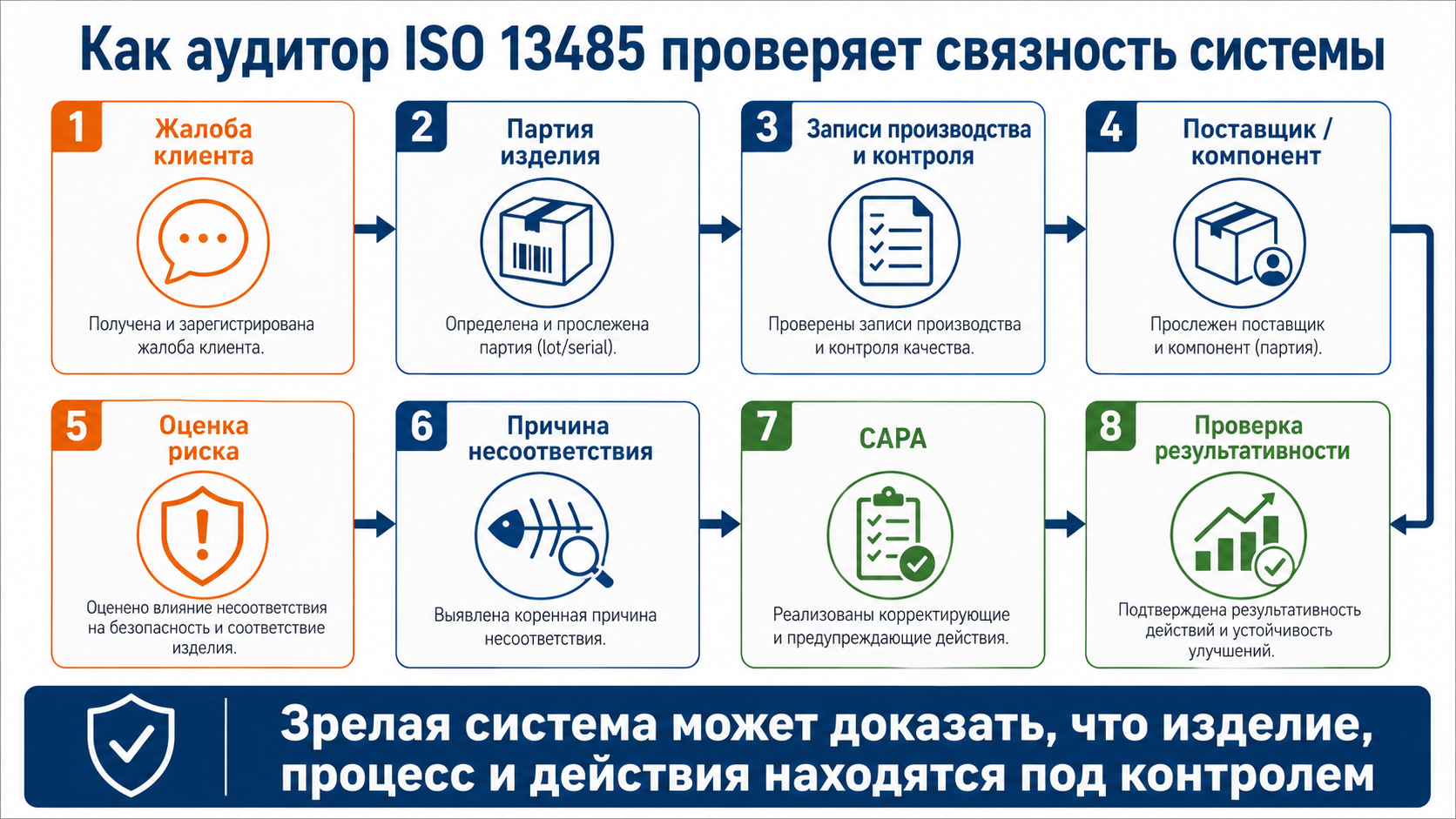
Аудит по ISO 13485 редко сводится к вопросу “есть ли у вас процедура”. Обычно аудитор проверяет, работает ли система в реальных процессах и можно ли это подтвердить документами и записями.
Как правило, внимание уделяют следующим вопросам:
- понимает ли руководство свою роль и ответственность;
- определены ли процессы и их взаимосвязи;
- как компания управляет рисками;
- как обеспечивается идентификация и прослеживаемость;
- как оцениваются и контролируются поставщики;
- какие процессы валидированы и почему;
- как оформляются и анализируются несоответствия;
- как работают корректирующие и предупреждающие действия;
- как обрабатываются жалобы и обратная связь;
- как управляются изменения;
- насколько записи полные, непротиворечивые и своевременные;
- соответствует ли реальная практика сотрудников утвержденным документам.
Хороший аудитор смотрит не на отдельные файлы, а на связность всей системы. Например, жалоба клиента может привести его к проверке истории партии, затем к конкретному поставщику, затем к оценке риска, затем к корректирующим действиям. Если элементы системы не связаны между собой, это быстро становится очевидно. Подробнее о том, какие вопросы может задавать аудитор и какие доказательства обычно смотрят на практике, смотри в материале «Аудит ISO 13485: какие вопросы задает аудитор».
Если компания рассматривает ISO 13485 не только как внутреннюю систему управления, но и как путь к официальному подтверждению соответствия, важно заранее понимать, как проходит сертификационный аудит, какие этапы включает проверка и сколько времени может занять подготовка. Подробнее об этом смотри в материале «Сертификация ISO 13485: как проходит аудит, этапы и сроки».
Практические рекомендации и лучшие подходы
Если компания только начинает внедрение ISO 13485, лучше идти не от шаблонов документов, а от реальных процессов и рисков. Сначала нужно понять, какие требования стандарта применимы к вашей деятельности, какие процессы критичны для безопасности и соответствия изделия, какие документы уже есть, а какие нужно разработать. Пошаговый подход к внедрению разобран в материале «Как внедрить ISO 13485: пошаговый план».
Сначала важно определить, чем именно занимается организация: разрабатывает продукцию, производит, стерилизует, упаковывает, обслуживает, продает, устанавливает или передает часть работ подрядчикам. От этого зависит логика системы и глубина отдельных требований.
Затем полезно построить карту процессов и понять, где находятся критические точки: входящие материалы, специальные процессы, маркировка, выпуск продукции, хранение, изменения, жалобы, возвраты и работа с поставщиками.
После этого стоит задать себе базовые вопросы:
- назначены ли владельцы процессов;
- какие записи подтверждают выполнение требований;
- где может потеряться прослеживаемость;
- какие процессы требуют валидации;
- как принимаются решения по отклонениям;
- как оценивается влияние изменений;
- как компания работает с жалобами и сигналами с рынка.
На практике особенно полезны такие шаги:
- Упростить документы до рабочего уровня.
- Документ должен помогать сотруднику выполнять работу правильно, а не существовать ради архива.
- Связать управление рисками с реальными решениями.
- Риски должны влиять на глубину контроля, частоту проверок, требования к поставщикам и объем валидации.
- Усилить контроль изменений.
- Нужен единый подход к оценке влияния изменений на изделие, процесс, документацию и обязательные требования.
- Сделать систему корректирующих действий действительно работающей.
- Не закрывать действия до тех пор, пока нет доказательств, что проблема устранена или существенно снижена.
- Отработать цепочку “жалоба — партия — поставщик — причина — действие”.
- Это один из лучших способов проверить зрелость системы.
- Готовить систему не к аудиту, а к устойчивой работе.
- Тогда и сертификация, и внешние проверки становятся следствием нормально выстроенного управления, а не разовой кампанией по подготовке.
Итоги
ISO 13485 — это не просто стандарт и не просто путь к сертификату. Это практическая модель управления качеством для компаний, работающих с медицинскими изделиями. Она помогает выстроить процессы так, чтобы изделие было не только произведено, но и находилось под контролем с точки зрения безопасности, соответствия требованиям, прослеживаемости и устойчивости бизнеса.
Для компании внедрение ISO 13485 означает переход от разрозненных действий к системной работе: с понятными ролями, записями, управлением рисками, контролем изменений, валидацией процессов, работой с жалобами и корректирующими действиями. Именно это делает систему полезной не только для аудитора, но и для самой организации.
Если говорить совсем просто, ISO 13485 отвечает на один ключевой вопрос: может ли компания стабильно и доказуемо выпускать медицинские изделия под контролем, а не “на опыте и старании сотрудников”. Если да, система менеджмента качества действительно работает. Если нет, сам по себе сертификат проблему не решит.
Другие материалы по теме ISO 13485
Если вы хотите глубже разобраться в отдельных требованиях стандарта, рекомендуем также посмотреть:
- Аудит ISO 13485: какие вопросы задает аудитор
- Жизненный цикл медицинского изделия и требования ISO 13485
- Политика и цели в области качества по ISO 13485
- Проектирование и разработка по ISO 13485: требования стандарта
- Управление поставщиками и закупками по ISO 13485
- Валидация процессов по ISO 13485: основные требования
- Прослеживаемость медицинских изделий по ISO 13485: что требует стандарт
- Управление рисками в ISO 13485: требования стандарта
- Документированная информация в ISO 13485: какие требования нужно учитывать
- ISO 13485 и ISO 14971: как связаны качество и управление рисками
- Сертификация ISO 13485: как проходит аудит, этапы и сроки
- Как внедрить ISO 13485: пошаговый план
- Какие документы нужны для ISO 13485
- ISO 13485 и ISO 9001: в чем разница
- Требования ISO 13485 — разбор по разделам и пунктам простыми словами
- Кому подходит ISO 13485 и зачем он нужен
Полезные сервисы и ресурсы по сертификации ISO
Планируете пройти сертификацию по ГОСТ ISO 13485 или другим стандартам? Отправьте одну заявку и получите до 5 коммерческих предложений за 1 раз только от аккредитованных ФСА органов по сертификации.
📢 Подписывайтесь на наш Telegram-канал с новостями и статьями по ISO;
💬 Присоединяйтесь к чату специалистов по системам менеджмента;
💰 Рассчитайте примерную стоимость сертификации в нашем онлайн-калькуляторе;
🗺️ Найдите орган по сертификации в нашем реестре — по стандарту или прямо на карте.
Планируете пройти сертификацию по ГОСТ ISO 13485 или другим стандартам? Отправьте одну заявку и получите до 5 коммерческих предложений за 1 раз только от аккредитованных ФСА органов по сертификации.
📢 Подписывайтесь на наш Telegram-канал с новостями и статьями по ISO;
💬 Присоединяйтесь к чату специалистов по системам менеджмента;
💰 Рассчитайте примерную стоимость сертификации в нашем онлайн-калькуляторе;
🗺️ Найдите орган по сертификации в нашем реестре — по стандарту или прямо на карте.